First post on here, apologies beforehand for any mistakes in etiquette. I should also say that I am very new to the field of light sheet microscopy, so please do correct me if anything I say is misguided.
P.S Apparently new users can only use 2 links and 1 embedded media per post, so where appropriate I have replaced the links with directions on where to access my images from my imgur account.
Background
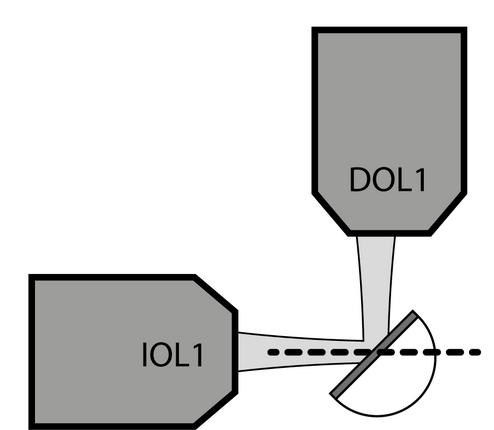
I have recently built a SPIM system based on the descSPIM open source microscope to be used for imaging cleared mouse organs (Brains and Pancreata mostly). It is essentially a compact Single Plane Illumination Microscope built entirely from parts commercially available from Thorlabs; a cylindrical lens is used for creating the light sheet. While the setup has gone well, and I have some “ok” preliminary data, I am consistently getting an axial resolution that is significantly more poor than what I believe I theoretically should achieve.
As I do not have a beam profiler, and have had trouble preparing a fluorescent bead calibration gel, I am basing my expectations on the in depth characterisation of the system in the original paper. In theory, I should be achieving a 3.45 micron lateral resolution, with the expected 3x decrease in axial resolution (~10 micron). As the first samples I imaged were prepared using (almost) the exact same protocol as the paper (CUBIC cleared nuclear stained mouse brains), I have some comparison images to illustrate my point.
The following is from the above linked paper; it is a PI stained mouse brain, which has been imaged in the dorsal-ventral plane (x-y), and shows the corresponding reoncstructed coronal section (x-z).
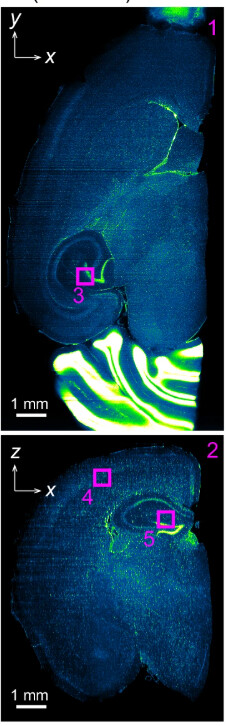
Here is from one of my prepared samples, the brain is damaged but should still illustrate the point. The only difference from this sample to the one above is I have used TO:PRO-1 instead of PI for the nuclear stain as my multi-line laser doesn’t have the wavelength capable of exciting PI.
X-Y

X-Z (from napari)

Thoughts so far:
My immediate first thought was that the cuvette holding my sample was undergoing some slight vibrations during the movement of the motor. However, when I imaged a nuclear stained pancreas in a PS cuvette fixed to my holder, and oversampled during imaging (i.e. moved the stage motor at a 10x slower speed to rule out miscalibration of the motor speed), I still had the same problem.
Secondly, I noticed that around certain features, for example the hippocampal region, there appears to be a sort of “doubling” or hazyness which I read in some papers can either be a result of poor clearing or an RI mismatch? As below…
[In above imgur account - image titled hippocampus doubling]
Does anyone have an intuition on which of the 2 is the more likely culprit (clearing or RI matching), and if they think this is the more probable cause of my axial resolution? If so, do you know what could be the possible cause of said problem and how I could fix it moving forward? The brain I presented was cleared for 6 days at 37 degrees in freshly made CUBIC-L so I thought the clearing should have been on the more extensive end. The RI matching was done for 2 days in freshly prepared CUBIC-R+. Here is what the agarose encased brain looked like in my cuvette:
[In above imgur account - image titled Cuvette - Optical Path]
Lastly, for preparing my agarose encased brains, I used a mould I designed and 3D printed out of PLA. As such, the mould has some very slight layer lines (printed with 0.1mm layer height) on the sides facing the direction of the excitation light path which are visible on the agarose after setting. The detection path is made using glass slides, thus have a very smooth finish. My thoughts were that as the agarose is resuspended in my RI matching medium, that the light path shouldn’t be refracted by these lines as the light first traverses through the glass → CUBIC-R+ interface. However, could this be what is causing my issues? I have tried smoothing out some of my gel moulds using Dichloro Methane, as it is very difficult to use sanding for molds that small, however I found that DCM is quite aggressive with melting PLA and in practice it is quite difficult to get a smooth finish. For what its worth, this is the view of the cuvette from the laser light path.
[In above imgur account - image titled Cuvette - Excitiation Path]
I would really appreciate some insight from people who have had more experience than me in preparing samples for SPIM microscopy, who may be able to guide me on how to troubleshoot my problems further!